








Metastasis en Gaia

Síndrome de abstinencia neonatal
El síndrome de abstinencia neonatal (SAN) es un grupo de problemas que pueden ocurrir cuando un bebé es expuesto a drogas opioides adictivas o medicamentos por un período de tiempo…

La campaña del gobierno de China para erradicar el pesimismo y las emociones negativas en redes sociales
Fuente: BBC. El gobierno de China ha puesto en su punto de mira una emoción que cree que se ha vuelto demasiado común en internet: la desesperanza. Esta semana, la…

Las fotos de la operación policial más letal de la historia de Río de Janeiro
Río de Janeiro vivió escenas de guerra durante una operación policial contra el narcotráfico que, según informaron autoridades locales, se convirtió en la más letal de la historia de la ciudad con…
Magnetismo

La NASA encuentra una inesperada relación entre la intensidad del campo magnético terrestre y los niveles de oxígeno
Fuente: Meteored. La capacidad de mantener vida en la Tierra podría ir más allá de tener agua y atmósfera. Un…
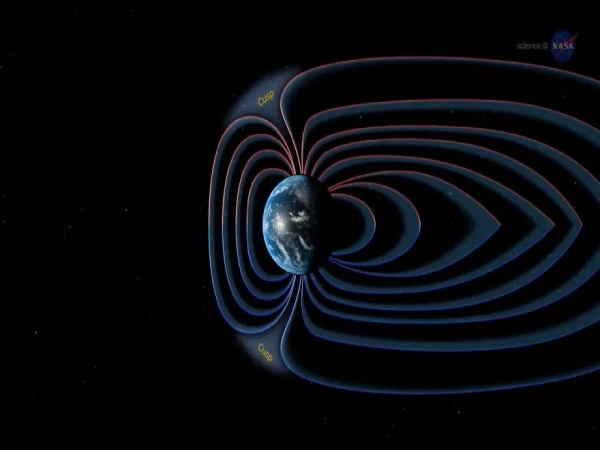
El campo magnético de la tierra se debilita, afecta a Argentina y pone en alerta a la NASA
Fuente: Río Negro. La Administración Nacional de Aeronáutica y el Espacio (NASA) advirtió sobre una alerta por una anomalía en el campo magnético…
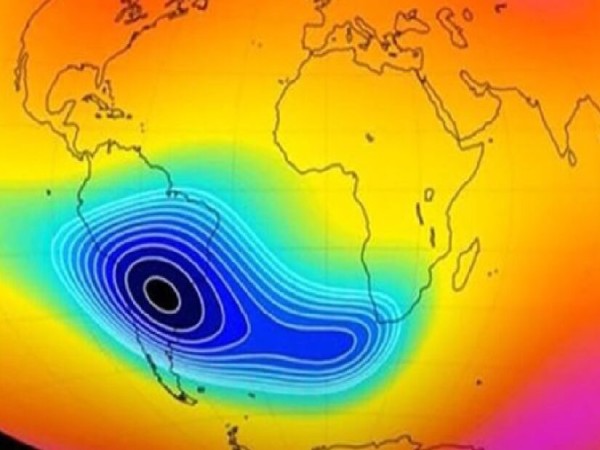
Qué es la «anomalía magnética»: el impacto en la Argentina por la alerta de la NASA
Fuente: El Dia. La Administración Nacional de Aeronáutica y el Espacio (NASA) monitorea la anomalía en el campo magnético de…

La física da un salto inesperado: descubren una conexión oculta entre magnetismo y vibraciones cristalinas
Fuente: Muyinteresante. Los descubrimientos científicos a menudo surgen de conexiones inesperadas entre conceptos que parecían no tener relación. Un equipo…
Ver más artículos sobre Magnetismo
Narcoterrorismo

Crimen organizado: el impacto geopolítico en la región
Fuente: Infobae La situación que el crimen organizado ha generado en el mundo es hiperdinámica, caótica, heterogénea y, por supuesto, lo suficientemente importante como para haber puesto en tensión el…
China creó una nueva cepa mutante de Covid-19 y temen que se convierta en una nueva pandemia
Fuente: El Cronista Un laboratorio chino ha creado una cepa mutante de coronavirus que ataca al cerebro de ratones. Según han indicado, esta tiene una tasa de mortalidad del 100%….

La historia del narcoterrorismo, desde las guerras del opio a los grupos yihadistas
Fuente: Lisa News El narcoterrorismo representa uno de los principales desafíos para la seguridad global y, desde el siglo XIX, impacta de diferentes formas y proporciones en todos los países…
Ver más artículos sobre Narcoterrorismo
Argenchina



En diciembre comenzará la producción de baterías de litio en el país
Fuente: Telam En el marco de la visita del presidente Alberto Fernández a las instalaciones de Y-TEC, en el partido bonaerense de Berisso, Roberto Salvarezza aseguró que el proyecto desplegado desde YPF apunta a un desarrollo “desde el salar hasta las baterías”. El presidente de Y-TEC (YPF-Tecnología), Roberto Salvarezza, afirmó este martes que en diciembre próximo se pondrá en marcha la producción…


Metahumano

La nueva tecnología que le da a los robots un cerebro hecho de células humanas
Fuente: El Confidencial. Científicos chinos han desarrollado un robot con un cerebro artificial creado en el laboratorio que, en combinación…

Crean robots con piel humana «viva» y el resultado es digno de una película de terror
Fuente: El Confidencial. Investigadores de la Universidad de Tokio han logrado este hito. No solo ayudará a conseguir robots humanoides…

Inteligencias artificiales que conversan con humanos empezaron a mostrar síntomas de ‘depresión’
Fuente: RPP Los chatbots de inteligencia artificial están asimilando la tristeza y el nihilismo de las personas con las que conversa. El estudio…

Humanidad

Cerebro

Más Noticias SOBRE
Humanidad
Coronavirus

Deshumanización


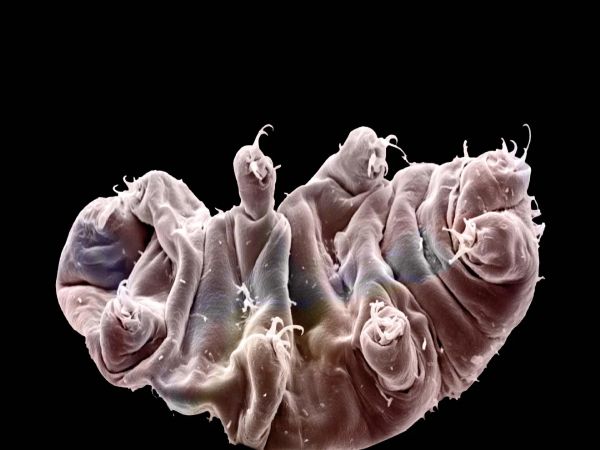
Una nueva especie de tardígrados confirma una habilidad especial para resistir la radiación extrema
Fuente: Wired. Los tardígrados, también conocidos como ‘osos de agua’, son pequeños animales invertebrados no mayores de un milímetro. Se caracterizan por una larguísima lista de habilidades que siguen fascinando a científicos y a otros especialistas; por ejemplo, pueden sobrevivir en condiciones ambientales extremas y son los animales más resistentes a las radiaciones ionizantes que conocemos. Estas criaturas soportan dosis de radiación…


Lo SOCIAL
Todos Necesitan Oír Esta Verdad Antes del Viernes — El Tiempo Se Agota | Elon Musk
Ver más artículos similares.

El movimiento solidario de Akamasoa Argentina
Fuente: Akamasoa. Akamasoa Argentina nace producto de un alma inquieta por cumplir un sueño contado a las hermanas del Padre…

Marco Aurelio, emperador romano: «Lo que no beneficia al enjambre, tampoco beneficia a la abeja»
Fuente: La Razón. Marco Aurelio Antonino, emperador del Imperio Romano entre los años 161 y 180 d.C., no solo fue…
La importancia del Olfato

Lo que los olores corporales revelan sobre tu estado de salud (y cómo pueden ayudar a diagnosticar enfermedades)
Fuente: BBC. Obviamente era una tontería. Así reaccionó la química analítica Perdita Barran cuando un compañero le habló de una…

Los perros perciben el estrés del ser humano
Fuente: Diario Hoy Psicólogos del Reino Unido pudieron confirmar que los perros son capaces de percibir el estrés en sus…

Sociología del olor
Fuente: Scielo CADA UNO DE NOSOTROS en todo momento, emitimos y percibimos olores, olemos y nos huelen, y tales olores…
Confieso que he Leído
Ana Herbsztein